|
|
"The people who are crazy enough to think they can change the world are the ones who do." |

Drug RepurposingProtein modeling, ligand binding site prediction, and sequence-order independent pocket matching are used to predict new protein targets, associated with rare diseases, for FDA approved drugs.
|

CheminformaticsInspired by LEGO, two software packages eMolFrag and eSynth computationally deconstruct chemical compounds into small fragments and build targeted libraries of new chemical compounds for virtual screening.
|
VirologyExperimental and computational projects to study Herpes viruses, find new protein targets to develop anti-viral drugs, and to investigate the combination of chemo and virotherapy to treat breast and prostate cancers.
|
Bioinspired STEM: Harness the wisdom generated in billions of years of evolution.
Drug Repositioning |
eModel-BDB & eRepo-ORP
- Naderi M, Brylinski M. Submitted. eModel-BDB: A database of comparative structure models of drug-target interactions from the Binding Database.
- Brylinski M, Naderi M, Govindaraj RG, Lemoine J. In Press. eRepo-ORP: Exploring the opportunity space to combat orphan diseases with existing drugs. J Mol Biol.
- Naderi M, Lemoine JM, Govindaraj RG, Kana OZ, Feinstein WP, Brylinski M. Submitted. Binding site matching in rational drug design: Algorithms and applications.
- Govindaraj RG, Naderi M, Lemoine J, Brylinski M. Submitted. Large-scale computational drug repositioning to find treatments for rare diseases.
By constructing protein models associated with rare diseases as well as protein-drug complexes that lack crystal structures computationally, we can predict and compare binding pockets in rare diseases and drug-bound protein complexes using high performance computers. The goal is to explored the possibility of repositioning FDA-approved drugs to treat orphan diseases.
We have so far explored the FDA-approved drugs curated in DrugBank, by generating the structure models of the target protein drug complexes as the reference binding pockets to find similar binding sites in proteins associated with rare diseases. We have one interesting candidate drug for an orphan condition that will be patented and published in the near future.
We have also compiled a larger dataset based on BindingDB, which includes 200k complexes of small molecules and proteins receptors. This dataset will be used as reference to compare protein binding sites associated with orphan diseases to find small molecule inhibitors. Although, not all rare diseases are cause by gain of function or cellular dysfunctions that can be treated with small molecule inhibitors.
The same protocol and datasets generated in this research, may also be used to further our understanding of the functions and relationships between proteins. Or, similar process can be used to inform crystallography efforts by predicting ligands that can stabilize a protein structure and promote crystal formation upon effective binding.


Cheminformatics |
eMolFrag & eSynth
- Naderi M, Alvin C, Ding Y, Mukhopadhyay S, Brylinski M. 2016. A graph-based approach to construct target-focused libraries for virtual screening. J Cheminform. 8:14.
- Liu T, Naderi M, Alvin C, Mukhopadhyay S, Brylinski M. 2017. Break down in order to build up: Decomposing small molecules for fragment-based drug design with eMolFrag. J Chem Inf Model. 57(4):627-631.
Think of this project as making LEGO pieces out of chemical molecules and then make whatever new molecule you can make with those LEGO pieces; all done in and by the computer! Together eMolFrag and eSynth do just that. We have devised algorithms for these two cheminformatics software tools for in silico drug design. eSynth is coded in C++. eMolFrag was initially programmed in bash using OpenBabel and small pieces of bash and perl snippets; the software is now fully coded in Python utilizing RDKit package, making it very easy to install and implement. By decomposing ligands of a certain protein(s) and reconstructing small molecules using these moieties following their original connectivity patterns we can build targeted virtual screening libraries of small molecules for specific protein receptors. The goal is to reduce the size of the chemical space to improve the virtual screening yield.
Primum non nocere: "First do no harm"
3. Pu L, Naderi M, Liu T, Wu HC, Mukhopadhyay S, Brylinski M. Submitted. eToxPred: A machine learning-based approach to estimate the toxicity of
drug candidates.
In order to improve the efficiency of our pipeline, we are developing filters that can identify and exclude molecules that do not match the profile of a drug-like compound. Following Lipinski rules, molecules are filtered based on their molecular weight, plogp, number of hydrogen bond acceptors and donors. But, as said in the Hippocratic Oath: first do no harm; therefore, we exclude chemicals that are predicted to be toxic. The prediction is made by a machine learning algorithm we developed, called eToxPred. Based on the available data for toxic and non-toxic chemical compounds, eToxPred has been taught to distinguish compounds that look like toxic compounds based on their chemical fingerprint.
drug candidates.
In order to improve the efficiency of our pipeline, we are developing filters that can identify and exclude molecules that do not match the profile of a drug-like compound. Following Lipinski rules, molecules are filtered based on their molecular weight, plogp, number of hydrogen bond acceptors and donors. But, as said in the Hippocratic Oath: first do no harm; therefore, we exclude chemicals that are predicted to be toxic. The prediction is made by a machine learning algorithm we developed, called eToxPred. Based on the available data for toxic and non-toxic chemical compounds, eToxPred has been taught to distinguish compounds that look like toxic compounds based on their chemical fingerprint.


Virology |
Computational virology
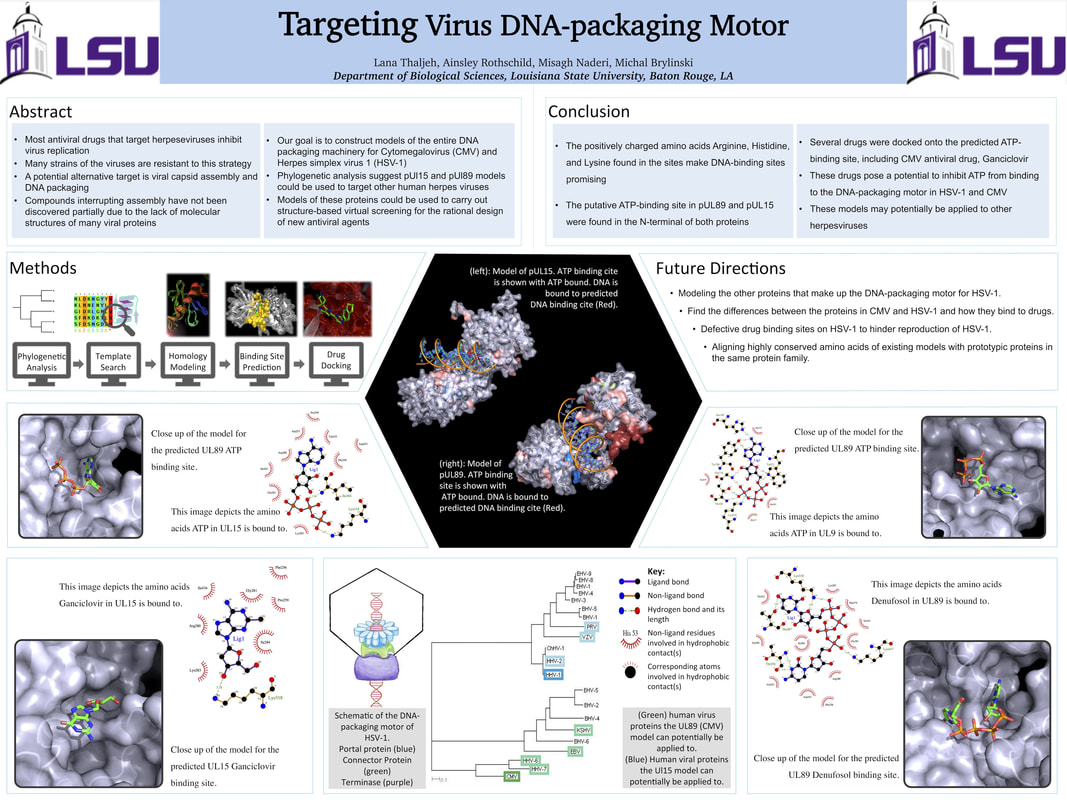
Antiviral drug discovery targeting DNA packaging motor
I am excited about this project, because it is my baby. I came up with the idea and currently am leading the project! And, if we find a drug I will be famous and probably rich! This thought keeps me going. Now, a brief explanation about the project.
To fight back with a viral infection, viruses can be stopped from getting into cell, once they are in the replication cycle can be stopped, viral proteins can be blocked to interfere with new viral particle formations, or the process loading the genetic material to a new virus capsid can be interrupted. The current antiviral drugs can be classified based on their mechanisms of interference with various stages of the virus lifecycle. The entry of the virus into host cells can be inhibited by interrupting the attachment (enfuvirtide), fusion (VIRIP), and uncoating of the virus (rimantadine). Another group of drugs interfere with viral replication and protein synthesis by inhibiting viral polymerases (acyclovir), nucleoside reverse transcriptases (tenofovis), and integrases (raltegravir). Drugs acting on viral spread (oseltamivir) prevent the release of virus particles from host cells. But, to my surprise there wasn't a drug that targets virus DNA packaging motor proteins specifically.
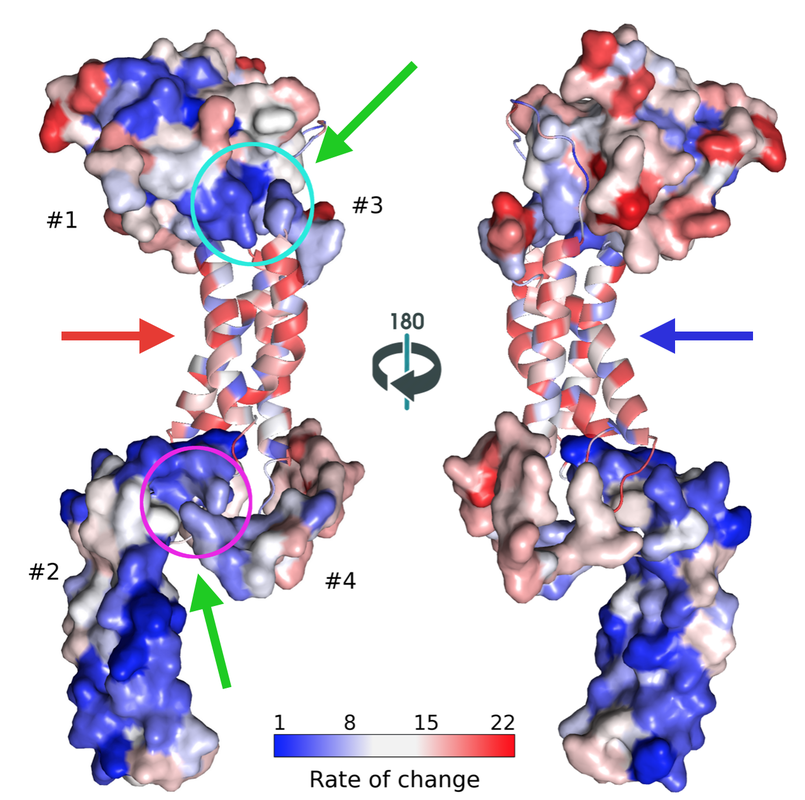
Our most interesting finding was that proteins involved in HSV-1 DNA packaging motor are amongst the most conserved based on sequence analysis. In contrast, DNA polymerases that are the most common targets for antiviral drugs have much higher rates of amino acid mutation. Thus, anti-DNA packaging drugs will be more difficult for the virus to be resistant against, which is what we need. And, that's what we are doing. We are modeling proteins in the motor to be able to find binding sites and possible initial lead compounds.
To fight back with a viral infection, viruses can be stopped from getting into cell, once they are in the replication cycle can be stopped, viral proteins can be blocked to interfere with new viral particle formations, or the process loading the genetic material to a new virus capsid can be interrupted. The current antiviral drugs can be classified based on their mechanisms of interference with various stages of the virus lifecycle. The entry of the virus into host cells can be inhibited by interrupting the attachment (enfuvirtide), fusion (VIRIP), and uncoating of the virus (rimantadine). Another group of drugs interfere with viral replication and protein synthesis by inhibiting viral polymerases (acyclovir), nucleoside reverse transcriptases (tenofovis), and integrases (raltegravir). Drugs acting on viral spread (oseltamivir) prevent the release of virus particles from host cells. But, to my surprise there wasn't a drug that targets virus DNA packaging motor proteins specifically.
Our most interesting finding was that proteins involved in HSV-1 DNA packaging motor are amongst the most conserved based on sequence analysis. In contrast, DNA polymerases that are the most common targets for antiviral drugs have much higher rates of amino acid mutation. Thus, anti-DNA packaging drugs will be more difficult for the virus to be resistant against, which is what we need. And, that's what we are doing. We are modeling proteins in the motor to be able to find binding sites and possible initial lead compounds.
|
Computational modeling of Herpes Simplex Virus proteins
- Chouljenko DV, Jambunathan N, Chouljenko VN, Naderi M, Brylinski M, Caskey JR, Kousoulas KG. 2016. Herpes simplex virus type 1 UL37 protein tyrosine residues conserved among all alphaherpesviruses are required for interactions with glycoprotein K (gK), cytoplasmic virion envelopment, and infectious virus production. J Virol. 90(22):10351-10361.
- Jambunathan N, Charles AS, Subramanian R, Saied AA, Naderi M, Rider P, Brylinski M, Chouljenko VN, Kousoulas KG. 2016. Deletion of a predicted β-sheet domain within the amino terminus of herpes simplex virus glycoprotein K conserved among alphaherpesviruses prevents virus entry into neuronal axons. J Virol. 90(5):2230-9.
- Rider P, Naderi M, Bergeron S, Chouljenko VN, Brylinski M, Kousoulas KG. 2017. Cysteines and N-glycosylation sites conserved among all alphaherpesviruses regulate membrane fusion in herpes simplex virus 1 infection. J Virol. 91(21)
After moving from the virology lab to the computational biology team, I reconsidered some of the common problems we were working on in virology in a new light. Sure enough, I came up with some ideas and ran them with my advisors and they liked them. That's how the computational virology projects started.
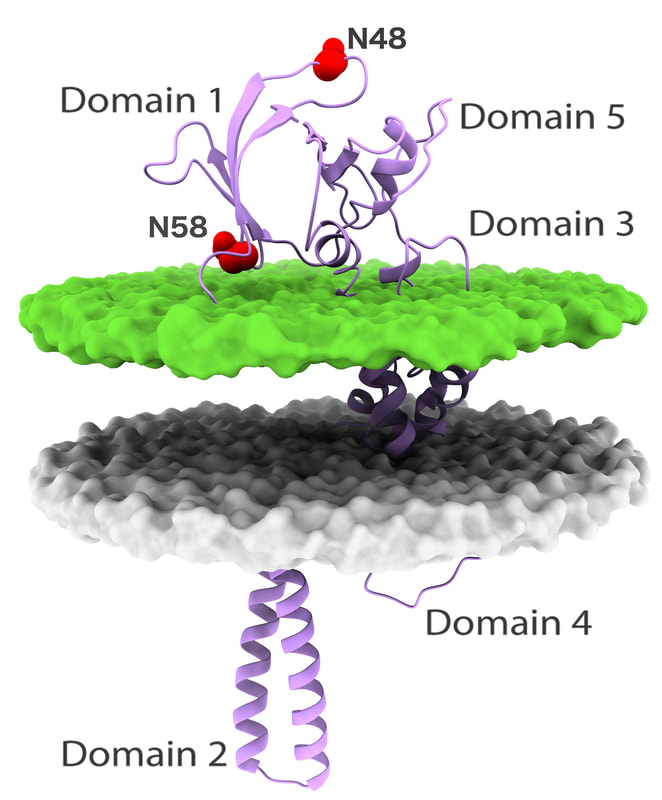
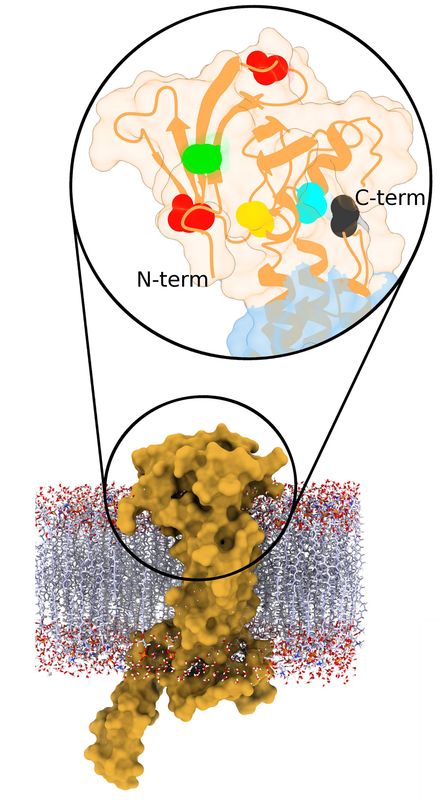
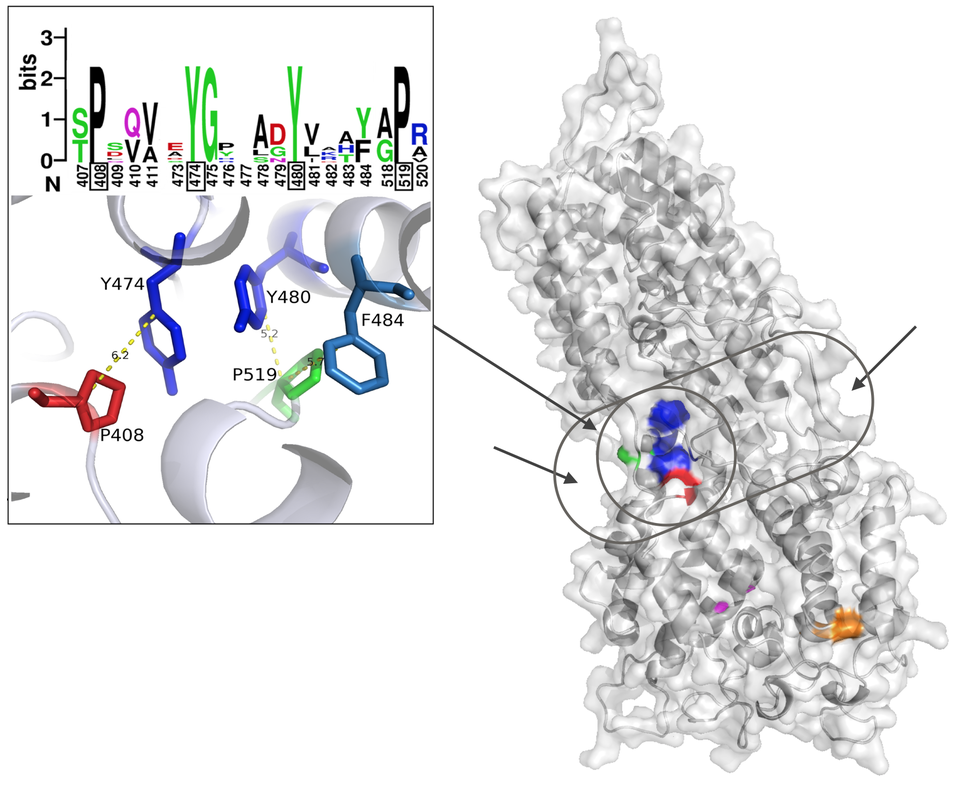
First we tackled the main project of the lab, which was Glycoprotein K in Herpes Simplex Virus (HSV). We proposed a quasi-3D model for the protein, but later on we constructed the first full model for this transmembrane protein. Based on the model we proposed multiple interesting domains and motifs; through mutation analysis performed by the virology lab we have so far generated very interesting insights. The most important finding so far is locating possible drug binding sites that we are doing more research on. The next step is to consider other protein partners of gK and model multi-protein machineries to understand how they work together.
Cancer virotherapy
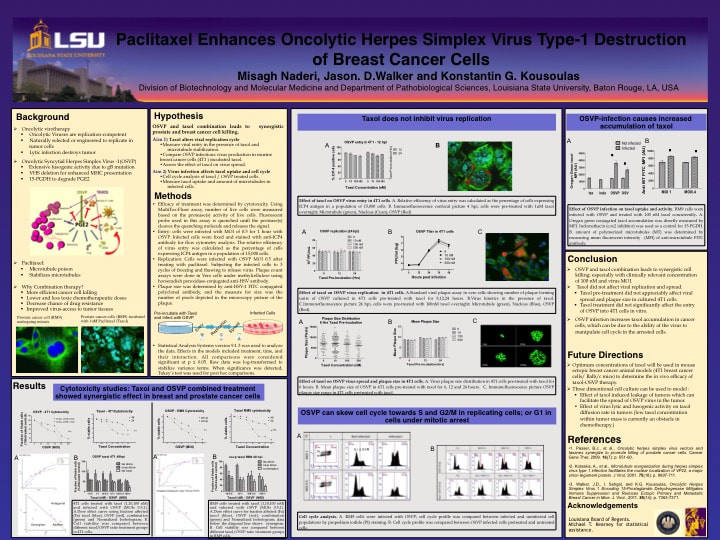
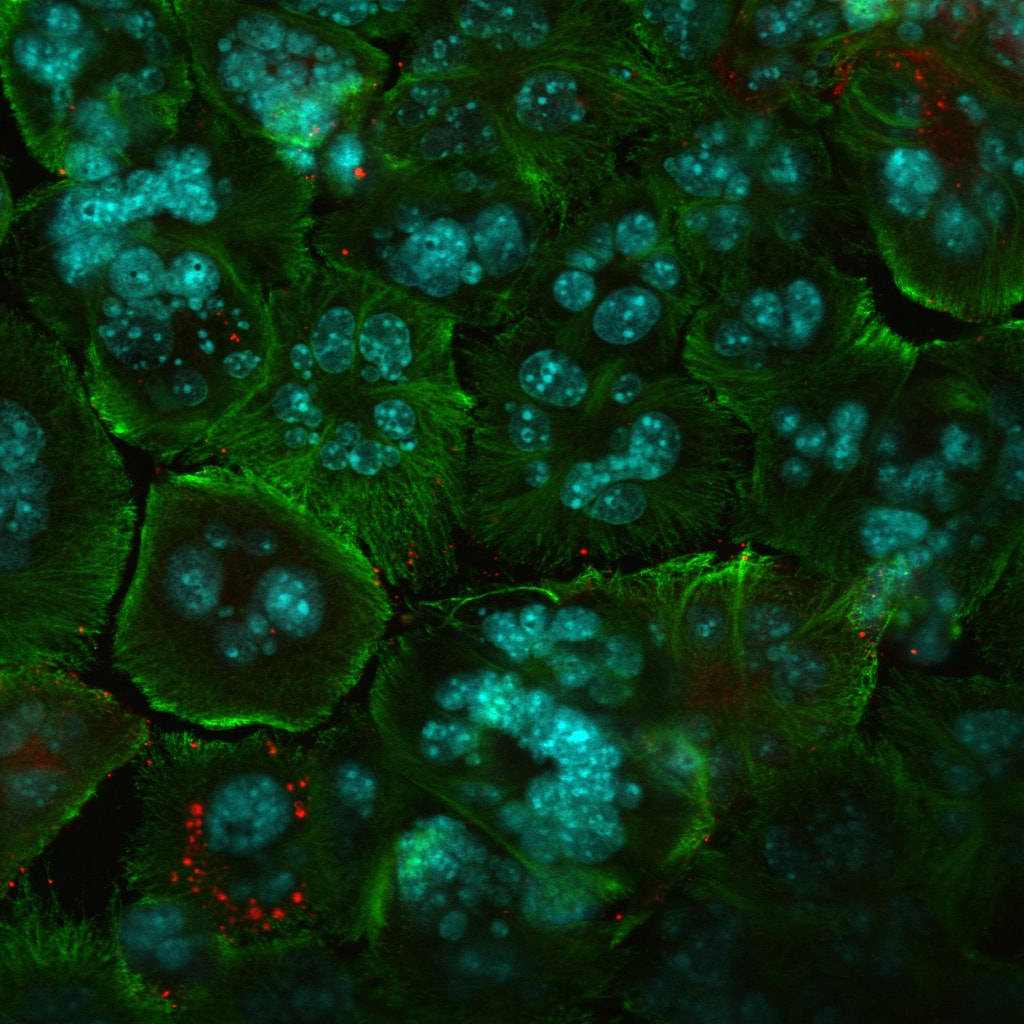
This is a molecular virology research that I performed in the wet-lab. Herpes simplex viruse was manipulated to attack and kill cancer cells. I studied the combination therapy using a specific strain of HSV together with chemotherapy to treat prostate and breast cancer. i investigated the effect of chemotherapy on virus life cycle and the effectiveness of the combination compared to each mode of therapy alone. The results showed that chemotherapy did not interfere with virus production and surprisingly increased the rate of virus entry into cells, and that the combination was synergistic. In other words, together virus and taxol worked better and killed more cancer cells than when used individually.